
Ο Ευρωπαϊκός Οργανισμός Φαρμάκων (ΕΜΑ) άνοιξε το δρόμο για την ταχεία αξιολόγηση ενός πειραματικού φαρμάκου, το οποίο αν εγκριθεί θα αποτελέσει την πρώτη θεραπεία, που αφορά τα αίτια της νόσου του Huntington. Πρόκειται για μία σπάνια και θανατηφόρα νευροεκφυλιστική νόσο.
Το φάρμακο φέρει την κωδική ονομασία RG6042 και έχει αναπτυχθεί από την ελβετική φαρμακοβιομηχανία Roche. Πρόσφατα, ο ΕΜΑ ενέταξε το RG6042 σε καθεστώς PRIME (PRIority MEdicines), το οποίο αφορά φάρμακα που μπορούν να προσφέρουν ένα σημαντικό θεραπευτικό πλεονέκτημα έναντι των υφιστάμενων θεραπειών ή να ωφελήσουν τους ασθενείς χωρίς θεραπευτικές επιλογές, παρέχοντας μια πορεία για την ταχύτερη αξιολόγηση από τον οργανισμό.
Οι δοκιμές
Η κίνηση αυτή βασίζεται κατά κύριο λόγο στα δεδομένα μιας διερευνητικής δοκιμής Φάσης Ι / ΙΙα του RG6042 που έδειξε σημαντική μείωση μίας πρωτεΐνης (mHTT), η οποία διασπά τα νευρικά κύτταρα στον εγκέφαλο. Η μελέτη έδειξε μια μέση μείωση κατά 40% (έως 60%) της συγκεκριμένης πρωτεΐνης στο εγκεφαλονωτιαίο υγρό των ενηλίκων ασθενών που έλαβαν RG6042 για τρεις μήνες στις δύο υψηλότερες δόσεις.
Επιπλέον, τα επίπεδα mHTT που μετρήθηκαν στο CSF εξακολουθούσαν να μειώνονται στην πλειονότητα των ασθενών που έλαβαν θεραπεία (περίπου 70%) από την τελευταία μέτρηση της μελέτης. Η Roche θα ξεκινήσει μια βασική μελέτη φάσης ΙΙΙ για την αξιολόγηση του RG6042 σε μεγαλύτερο πληθυσμό ασθενών για να καθορίσει περαιτέρω το προφίλ ασφάλειας και να καθορίσει εάν μπορεί να επιβραδύνει τη νόσο.
Η νόσος
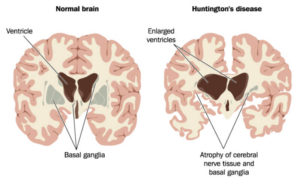
H νόσος του Huntington αποτελεί γενετική κληρονομική νευροεκφυλιστική διαταραχή, η οποία επηρεάζει σταδιακά τη φυσική, γνωστική και συναισθηματική κατάσταση του ατόμου που νοσεί. Στις περισσότερες περιπτώσεις, τα πρώτα συμπτώματα εκδηλώνονται σε ηλικία μεταξύ των 30 και 50 ετών.
Η νόσος προκαλείται από μετάλλαξη στο γονίδιο της «χαντινγκτίνης» (γονίδιο ΙΤ-15 ή HD ή HTT), στο χρωμόσωμα 4, το οποίο φέρει φυσιολογικά κάθε άνθρωπος. Κληρονομείται με αυτοσωματικό επικρατητικό τρόπο. Αυτό σημαίνει ότι, εφόσον ένας από τους δύο γονείς φέρει το μεταλλαγμένο γονίδιο, τότε κάθε παιδί, αγόρι ή κορίτσι, έχει ίσες πιθανότητες (50%) να κληρονομήσει ή να μην κληρονομήσει τη νόσο.
Το φυσιολογικό γονίδιο της «χαντινγκτίνης» περιέχει κανονικά 10-35 επαναλήψεις CAG (κυτοσίνη – αδενίνη – γουανίνη). Ενώ η αύξηση του αριθμού των επαναλήψεων ως τις 27-35 θεωρείται εντός των ανώτερων φυσιολογικών ορίων και δεν προξενεί τη νόσο, υπάρχει, παρόλα αυτά, πιθανότητα για ανάπτυξη της Νόσου στις επόμενες γενιές. Άτομα με αύξηση των επαναλήψεων μεταξύ 36-39 έχουν μειωμένη πιθανότητα να αναπτύξουν τη Νόσο. Κάποιοι θα αναπτύξουν συμπτώματα, ενώ άλλοι όχι.
Άτομα με 40 ή περισσότερες επαναλήψεις προβλέπεται πως θα αναπτύξουν τη νόσο κάποια στιγμή στη ζωή τους. Σύμφωνα με έρευνες, όσο μικρότερος είναι ο αριθμός των επαναλήψεων, τόσο αργότερα θα εμφανιστούν τα πρώτα συμπτώματα. Ο αυξημένος αριθμός επαναλήψεων CAG στο μεταλλαγμένο γονίδιο έχει ως αποτέλεσμα την παραγωγή παθολογικής πρωτεΐνης χαντινγκτίνης, η οποία είναι επιβλαβής για τα νευρικά κύτταρα.
Τα συμπτώματα της νόσου διακρίνονται σε νοητικά και κινητικά. Συνήθως η νόσος αρχίζει με διαταραχές από την ψυχική σφαίρα: ευερθιστότητα, καχυποψία, μερικές φορές θρησκοληψία και μειωμένο αυτοέλεγχο. Συχνά εμφανίζεται αλκοολισμός, διαταραχές του συναισθήματος και κοινωνική απόσυρση και στα προχωρημένα στάδια παραισθήσεις και ψευδαισθήσεις
Τα κινητικά συμπτώματα στα πρώτα στάδια της νόσου είναι ηπιότερα και συνίστανται σε επιβράδυνση των κινήσεων των δακτύλων και των άκρων γενικότερα και ο μυϊκός τόνος είναι κατά κανόνα μειωμένος. Ο λόγος μοιάζει αταξικός, ενώ έντονες είναι οι αυτόματες κινήσεις της γλώσσας και της περιστοματικής περιοχής.












Comments (0)